Plots a 2d binned histogram plot from two given bigWig files
Usage
plot_bw_bins_density(
x,
y,
bg_x = NULL,
bg_y = NULL,
norm_mode_x = "fc",
norm_mode_y = "fc",
bin_size = 10000,
genome = "mm9",
plot_binwidth = 0.05,
highlight = NULL,
remove_top = 0,
verbose = TRUE,
selection = NULL,
default_na = NA_real_,
scaling = "none",
canonical = FALSE
)Arguments
- x
BigWig file corresponding to the x axis.
- y
BigWig file corresponding to the y axis.
- bg_x
BigWig file to be used as x axis background (us. input).
- bg_y
BigWig file to be used as y axis background (us. input).
- norm_mode_x
Normalization mode for x axis.
- norm_mode_y
Normalization mode for y axis.
- bin_size
Bin size.
- genome
Genome. Available choices are mm9, hg38.
- plot_binwidth
Resolution of the bins in the density histogram (different to genomic bin size)
- highlight
List of bed files to use as highlight for subgroups.
- remove_top
Return range 0-(1-remove_top). By default returns the whole distribution (remove_top == 0).
- verbose
Put a caption with relevant parameters on the plot.
- selection
A GRanges object to restrict binning to a certain set of intervals. This is useful for debugging and improving performance of locus specific analyses.
- default_na
Default value for missing values
- scaling
If none, no operation is performed (default). If relative, values are divided by global mean (1x genome coverage).
- canonical
Use only canonical chromosomes (default: FALSE)
Details
Values in x and y axis can be normalized using background bigWig files (usually input files). By default, the value shown will be x / bg_x per bin. If norm_func_x or norm_func_y are provided, this can be changed to any given function, for instance, if norm_func_x = log2, values on the x axis will represent log2(x / bg_x) for each bin.
Values that are invalid (NaN, Inf, -Inf) in doing such normalization will be ignored and shown as warnings, as this is ggplot default behavior.
Examples
# Get the raw files
bw <- system.file("extdata", "sample_H33_ChIP.bw", package="wigglescout")
bw2 <- system.file("extdata",
"sample_H3K9me3_ChIP.bw", package="wigglescout")
bed <- system.file("extdata", "sample_genes_mm9.bed", package="wigglescout")
# Sample bigWig files only have valid values on this region
locus <- GenomicRanges::GRanges(
seqnames = "chr15",
IRanges::IRanges(102600000, 103100000)
)
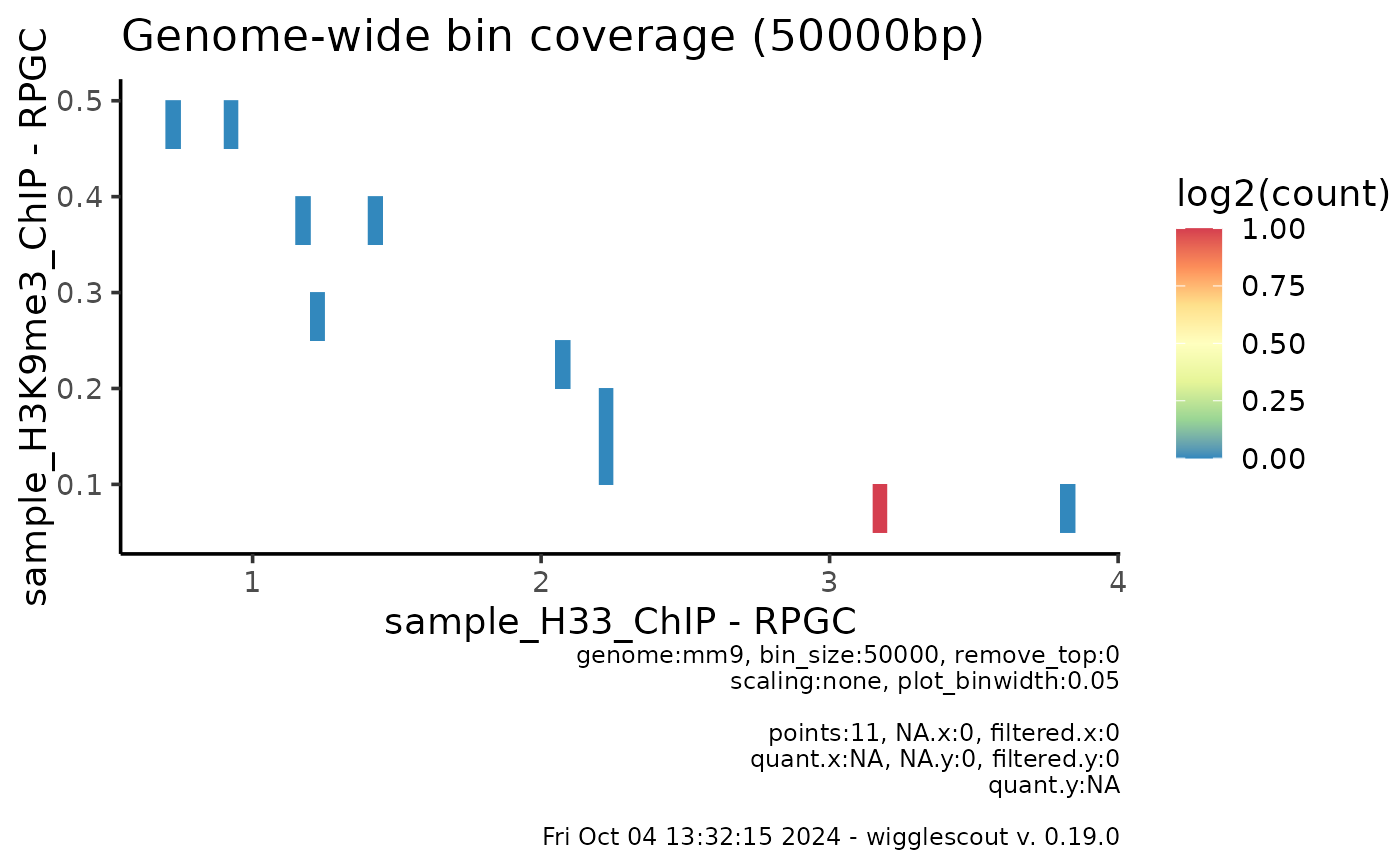
plot_bw_bins_density(bw, bw2, bin_size = 50000, selection = locus)
#> Warning: Not all bigWig files or the genome tiling provided share the same sequence info.
#> Common to all (1): chr15
#> Missing in some of the files: chr1, chr2, chr3, chr4, chr5 ...
#> This can be due to different versions of the same reference genome, or to completely different organisms. Make sure these match!
#> Warning: Not all bigWig files or the genome tiling provided share the same sequence info.
#> Common to all (1): chr15
#> Missing in some of the files: chr1, chr2, chr3, chr4, chr5 ...
#> This can be due to different versions of the same reference genome, or to completely different organisms. Make sure these match!
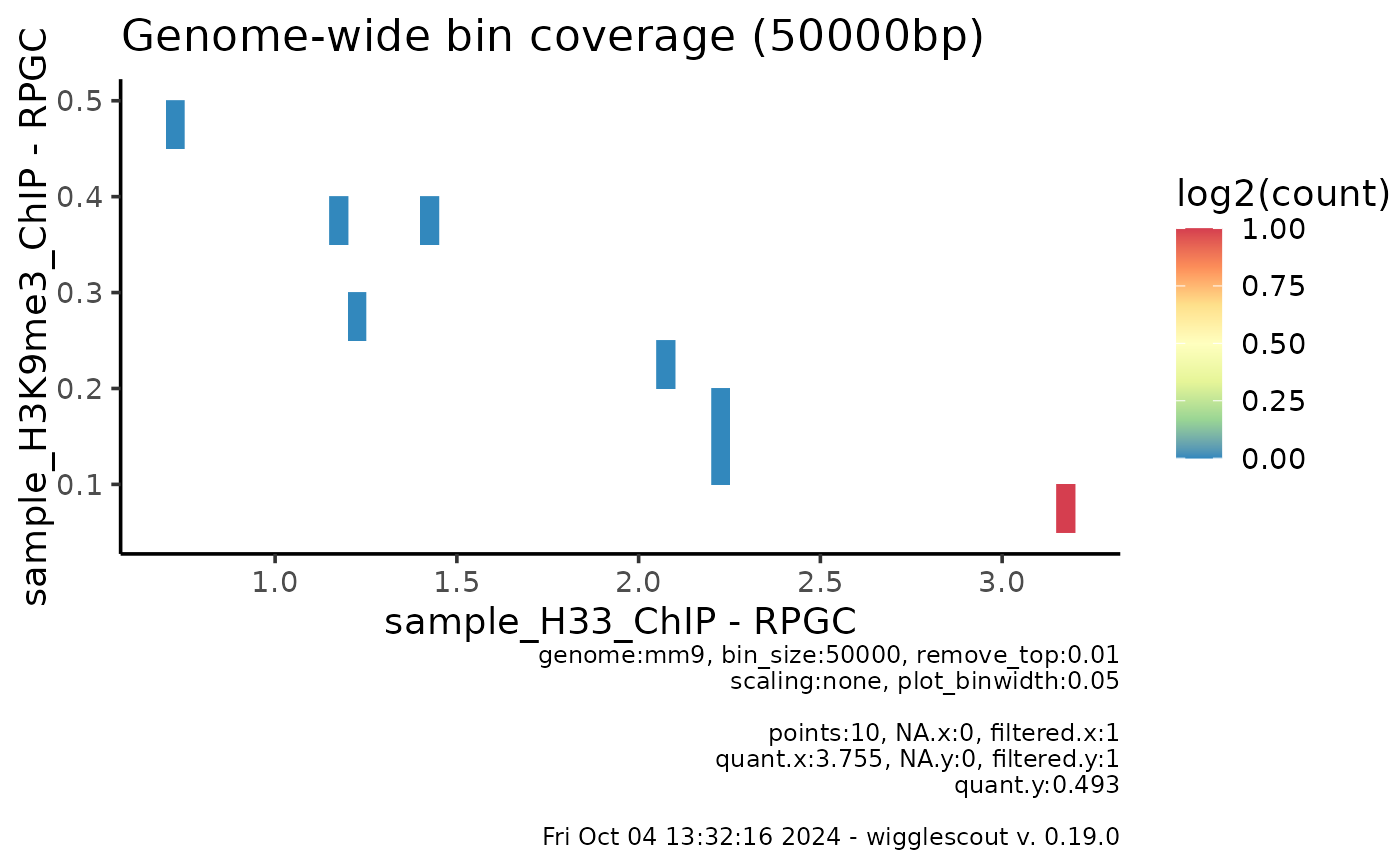
 plot_bw_bins_density(bw, bw2, bin_size = 50000, selection = locus,
remove_top = 0.01)
#> Warning: Not all bigWig files or the genome tiling provided share the same sequence info.
#> Common to all (1): chr15
#> Missing in some of the files: chr1, chr2, chr3, chr4, chr5 ...
#> This can be due to different versions of the same reference genome, or to completely different organisms. Make sure these match!
#> Warning: Not all bigWig files or the genome tiling provided share the same sequence info.
#> Common to all (1): chr15
#> Missing in some of the files: chr1, chr2, chr3, chr4, chr5 ...
#> This can be due to different versions of the same reference genome, or to completely different organisms. Make sure these match!
#> Warning: Removed 1 row containing non-finite outside the scale range (`stat_bin2d()`).
plot_bw_bins_density(bw, bw2, bin_size = 50000, selection = locus,
remove_top = 0.01)
#> Warning: Not all bigWig files or the genome tiling provided share the same sequence info.
#> Common to all (1): chr15
#> Missing in some of the files: chr1, chr2, chr3, chr4, chr5 ...
#> This can be due to different versions of the same reference genome, or to completely different organisms. Make sure these match!
#> Warning: Not all bigWig files or the genome tiling provided share the same sequence info.
#> Common to all (1): chr15
#> Missing in some of the files: chr1, chr2, chr3, chr4, chr5 ...
#> This can be due to different versions of the same reference genome, or to completely different organisms. Make sure these match!
#> Warning: Removed 1 row containing non-finite outside the scale range (`stat_bin2d()`).